Effect of Jaeumkanghwatang (JEKHT), a Polyherbal Formula on the Pharmacokinetics Profiles of Tamoxifen in Male SD Rats (1) - Single Oral Combination Treatment of Tamoxifen 50 mg/kg with JEKHT 100 mg/kg within 5 min -
Article information
Abstract
Objectives
The objective of this study was to elucidate the effect of Jaeumkanghwatang (JEKHT) on the plasma concentration and pharmacokinetics of tamoxifen in combination therapy as a process of the comprehensive and integrative medicine against breast cancer.
Methods
After 50 mg/kg of tamoxifen treatment, JEKHT 100 mg/kg was orally administered within 5 min. The plasma were collected at 30 min before administration, 30min, 1, 2, 3, 4, 6, 8 and 24 hrs after end of JEKHT treatment, and plasma concentrations of tamoxifen were analyzed using LC-MS/MS methods. PK parameters of tamoxifen (Tmax, Cmax, AUC, t1/2 and MRTinf) were analysis as compared with tamoxifen single administered rats.
Results
JEKHT did not influenced on the plasma concentrations and pharmacokinetics of tamoxifen after single oral co-administration, within 5min except for some negligible effects on plasma concentration. The Tmax, Cmax, AUC, t1/2 and MRTinf of tamoxifen in co-administered rats were quite similar to those of tamoxifen single treated rats.
Conclusions
Based on the results of the present study, JEKHT did not influenced on the oral bioavailability of tamoxifen, when they were single co-administered within 5min. However, more detail pharmacokinetic studies should be tested to conclude the possibilities that can be used as comprehensive and integrative therapy with JEKHT and tamoxifen for breast cancers, when they were co-administered, like the effects on the pretreatment of JEKHT and after repeat co-administrations.
Introduction
Tamoxifen (Nolvadex™) is a nonsteroidal estrogen agonist-antagonist antineoplastic agent has been used for breast cancer1). It is the usual endocrine (anti-estrogen) therapy for hormone receptor-positive breast cancer in pre-menopausal women, and is also a standard in post-menopausal women although aromatase inhibitors are also frequently used in that setting2,3). In addition, tamoxifen also used to treat infertility in women with anovulatory disorders4,5) and prevention for gynecomastia6,7) and bipolar disorder8,9) as anti-angiogenesis10), control of gene expression11), and treat Riedel thyroiditis12,13) and Albright’s syndrome14,15). Tamoxifen competitively binds to estrogen receptors on tumors and other tissue targets, producing a nuclear complex that decreases DNA synthesis and inhibits estrogen effects. It is a nonsteroidal agent with potent antiestrogenic properties which compete with estrogen for binding sites in breast and other tissues. Tamoxifen causes cells to remain in the G0 and G1 phases of the cell cycle. Because it prevents (pre)cancerous cells from dividing but does not cause cell death, tamoxifen is cytostatic rather than cytocidal16–18). However, various side effects related to tamoxifen treatment also have been arise as bone loss in premenopausal women who continue to menstruate after adjuvant chemotherapy19), endometrial changes, including cancer, are among tamoxifen’s side effects20), increased risk of thromboembolism21), cause of fatty liver22), reduced cognition23), semantic memory scores24) and libido25,26), and premature growth plate fusion27). Tamoxifen also depress the immune response28,29), and it also known that hypersensitivity to tamoxifen or any ingredient in the formulation 30,31).
As results of combination therapies with other drugs to improve the side effects of tamoxifen or to achieve synergic effects, various drug-drug interactions of tamoxifen have been evaluated; Because tamoxifen was metabolized by a substrate of CYP3A, 2C9, 2D632,33), it interacted with various drugs, namely, combinations containing any of the following medications, depending on the amount present, may also interact with aminoglutethimide - decreased plasma tamoxifen and N-desmethyltamoxifen concentrations34), anticoagulants - enhanced warfarin effects35,36), bromocriptine - increased plasma tamoxifen and N-desmethyltamoxifen concentrations37), letrozole - decreased plasma letrozole concentrationsa38), medroxyprogesterone - decreased plasma N-desmethyltamoxifen concentrations but did not reduce plasma tamoxifen concentrations39), phenobarbital - decreased plasma tamoxifen concentrations40), rifampin - decreased plasma tamoxifen and N-desmethyltamoxifen concentrations41), and cyclosporine, erythromycin, diltiazem, erythromycin and nifedipine – competitively inhibited formation of N-desmethyltamoxifen in vitro42–44), respectively. However, interactions with herbal products have not been established except for some restricted natural compounds; tamoxifen enhanced warfarin effects, and it is contraindicate that co-administration of tamoxifen and wafarin35,36).
Jaeumkanghwatang (JEKHT) is a traditional yin-tonifying herbal medicine has been used for various oriental obstetrical and gynecological fields and it comprises of 12 kinds of herbs like Glycyrrhizae Radix et Rhizoma, Angelicae Gigantis Radix, Zizyphi Fructus, Liriopis Tuber, Atractylodis Rhizoma Alba, Paeoniae Radix, Anemarrhenae Rhizoma, Rehmanniae Radix Crudus, Citri Unshii Pericarpium, Phellodendri Cortex, Zingiberis Rhizoma Crudus and Asparagi Tuber45) (Table 1). It is widely used in China, Japan, and Korea to treat bronchitis and tuberculosis46) with in some immune stimulation effects45). JEKHT has been demonstrated anti-allergic properties in vitro study and they include suppression of secretion of inflammatory cytokines through blockade of NF-κb activation47). In addition, It has been reported that JEKHT has beneficial effects in the treatment of patients with bronchial asthma46) and the relieving hot flash with JEKHT, representative side effect in tamoxifen treated patients with breast cancer, was reported recently48).
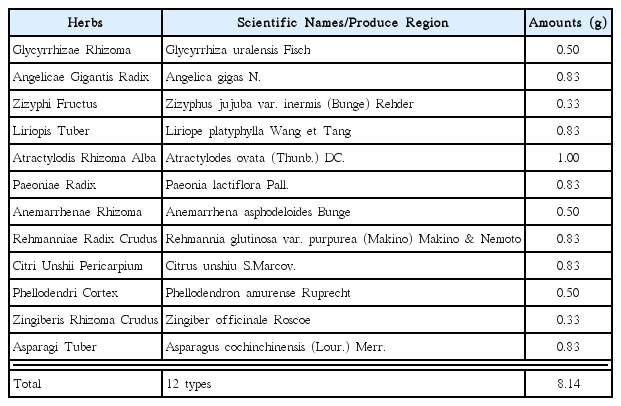
Composition of JEKHT Used in This Study
In the present study, the effects of JEKHT co-administration on the plasma concentration and pharmacokinetics of tamoxifen were observed as a process of the comprehensive and integrative medicine, against breast cancers.
Materials & methods
1. Animals and husbandry
Total ten male Sprague-Dawley (SD) rats (6-wk old upon receipt, SLC, Japan) were used after acclimatization for 16 days. Animals were allocated five per polycarbonate cage in a temperature (20–25 °C) and humidity (40–45%) controlled room. Light : dark cycle was 12 hr : 12 hr and feed (Samyang, Korea) and water were supplied free to access. All animals were marked by picric acid, and overnight fasted (about 18 hrs; water was not restricted) before treatment, and further fasted during 3 hrs after end of treatment. All laboratory animals were treated according to the national regulations of the usage and welfare of laboratory animals, and approved by the Institutional Animal Care and Use Committee in Daegu Haany University (Gyeongsan, Gyeongbuk, Korea) prior to animal experiment.
2. Test articles and formulation
JEKHT, prepared and purchase from Hanzung Pharm. Co. (Daejeon, Korea), and tamoxifen (Hangzhou Tacon Co., Ltd, Hangzhou, China) was used as control drug. Individual compositions of 12 kinds of herbs in JEKHT were listed in Table 1. Tamoxifen and powders of JEKHT extracts were stored in a refrigerator at 4 °C to protect from light and degeneration until use. Both drugs are well dissolved (up to 20 mg/ml solutions in JEKHT and up to 10 mg/ml solutions in tamoxifen) in distilled water as vehicle, respectively.
3. Groupings and administration
Five rats per group (two groups) were used in this study. The doses of test materials were selected based on their toxicity and pharmacodynamics – 50 mg/kg of tamoxifen with 100 mg/kg of JEKHT. After 50 mg/kg of tamoxifen treatment, JEKHT 100 mg/kg was orally administered, within 5 min. In tamoxifen single treated rats, 50 mg/kg of tamoxifen was orally administered and 5 min after treatment, only distilled water 5 ml/kg was orally administered, instead of JEKHT solutions. Each tamoxifen or JEKHT was single orally administered, in a volume of 5 ml/kg, dissolved in distilled water.
4. Plasma collections
All rats were slightly anesthesia under ethyl ether (Duksan Pure Chemical, Seoul, Korea) and blood samples (0.5 ml) were collected into 50IU heparinized tubes via the orbital plexus at 30 min before treatment (as a control), 30 min, 1, 2, 3, 4, 6, 8 and 24 hrs after end of oral administration. Blood samples were immediately centrifuged for 10 min at 13,000 rpm and about 0.3 ml aliquots of plasma were stored in a −70 °C deep freezer until analysis of tamoxifen.
5. Sample preparation and calibrations
Primary stock solution, 1.0 mg/ml of tamoxifen in 50% acetonitrile (Sigma, MO, USA) mixtures with distilled water and internal standard working solution, carbamazepine (Sigma, MO, USA) 500 ng/ml in acetonitrile were prepared. Working standard solutions were prepared by dilution with acetonitrile. All standard solutions were stored at −20 °C in the dark when not in use, and calibrated the standard samples as 100 μl of blank plasma, working standard solutions and internal standard working solution were mixed with 100 μl of acetonitrile. The mixtures were mixed by vortex-mixing and centrifuged at 12,000 rpm for 10 min at 4 °C. The clear supernatants were transferred to injection vials and the aliquot was injected into the LC-MS/MS system. In addition, 100 μl of sample plasma and internal standard working solution were mixed with 200 μl of acetonitrile. The mixtures were mixed by vortex-mixing and centrifuged at 12,000 rpm for 10 min at 4 °C. Clear supernatants (5.0 μl) were directly transferred to injection vials and the aliquot was injected into the LC-MS/MS system.
6. LC-MS/MS conditions
Concentrations of tamoxifen in the rat plasma samples were determined LC-MS/MS method. Chromatographic analysis was performed using an Agilent 1100 Series HPLC (Agilent Technologies, CA, USA) equipped with on-line degasser, binary pump, autosampler and column compartment. Separation of the analyte from potentially interfering material was achieved at ambient temperature using Waters Xterra MS C18 columns (2.1×50 mm, 3.5 μ m) (Waters Corp., MA, USA) at column oven 30 °C. The mobile phase used for the chromatographic separation was composed of 5% acetonitrile/95% distilled water (0.1% formic acid) to 95% acetonitrile/5% distilled water (0.1% formic acid), and was delivered isocratically at a flow rate of 0.35 ml/min. The column effluent was monitored using an API 2000 triple-quadruple mass-spectometric detector (Applied Biosystems, CA, USA). The instrument was equipped with an electrospray interface in positive ion mode, and controlled by the Analyst version 1.4.2 software (Applied Biosystems, CA, USA). Samples were introduced to the interface through a Turbo IonSpray with the temperature set at 400 °C. A high positive voltage of 5.0 kV was applied to the ion spray. Nitrogen was used as the nebulizer gas, curtain gas, and collision gas with the settings of 12, 6, and 8, respectively. The multiple reaction monitoring (MRM) detection method was employed for the detection of tamoxifen; the transitions monitored were carbamazepine (IS): m/z 237>194 (Retention time: 2.4 min), tamoxifen: 372>72 (Retention time: 2.3 min). Calibration curves of tamoxifen were linear over the ranges studied with r2>0.999. The lower limit of quantification of the tamoxifen in the rat plasma was 1 ng/ml.
7. Pharmacokinetic analysis
The plasma concentration data were analyzed using a noncompartmental method on commercial pharmacokinetics data analyzer programs (PK solutions 2.0; Summit, CO, USA)49,50). The elimination rate constant (Kel) was calculated by the log-linear regression of tamoxifen concentration data during the elimination phase, and the terminal half-life (t1/2) was calculated by 0.693/Kel. The peak concentration (Cmax) and time to reach the peak concentration (Tmax) of tamoxifen in the plasma were obtained by visual inspection of the data in the concentration-time curve. The area under the plasma concentration-time curve (AUC0-t) from time zero to the time of the last measured concentration (Clast) was calculated using the linear trapezoidal rule 51). The AUC zero to infinity (AUC0-inf) was obtained by adding AUC0-t and the extrapolated area was determined by Clast/Kel. The mean residence time infinity (MRTinf) was calculated by dividing the first moment of AUC (AUMC0-inf) by AUC0-inf.
8. Statistical analyses
All the means are presented with their standard deviation of five rats (Mean ± SD of five rat plasma concentrations of tamoxifen). The pharmacokinetic parameters were compared using a non-parametric comparison test, Mann-Whitney U (MW) test, on the SPSS for Windows (Release 14.0K, SPSS Inc., USA). A p-value <0.05 was considered statistically significant. In addition, the percent changes between tamoxifen single treated rats and tamoxifen with JEKHT co-administered rats were calculated to help the understanding of the effects of co-administration.
Results
1. Changes on the plasma concentrations of tamoxifen
Tamoxifen was detected from 30 min to 24 hrs after end of administration in the both tamoxifen single and co-administered rats with JEKHT, respectively. No meaningful and no-significant changes on the plasma tamoxifen concentrations were detected in JEKHT co-administered rats as compared with tamoxifen single treated rats after single co-administration of tamoxifen 50 mg/kg with JEKHT 100mg/kg, except for non-significant slight increases of plasma tamoxifen levels detected at 30 min and 1 hr after administration in JEKHT co-administered rats as compared with tamoxifen single treated rats (Fig 1). The plasma tamoxifen concentrations at 30 min, 1, 2, 3, 4, 6, 8 and 24 hrs after end of treatment were changed as 26.53, 49.94, −6.49, −3.76, −6.59, −13.04, −15.36 and −8.81% in tamoxifen + JEKHT treated rats as compared with tamoxifen single treated rats, respectively.
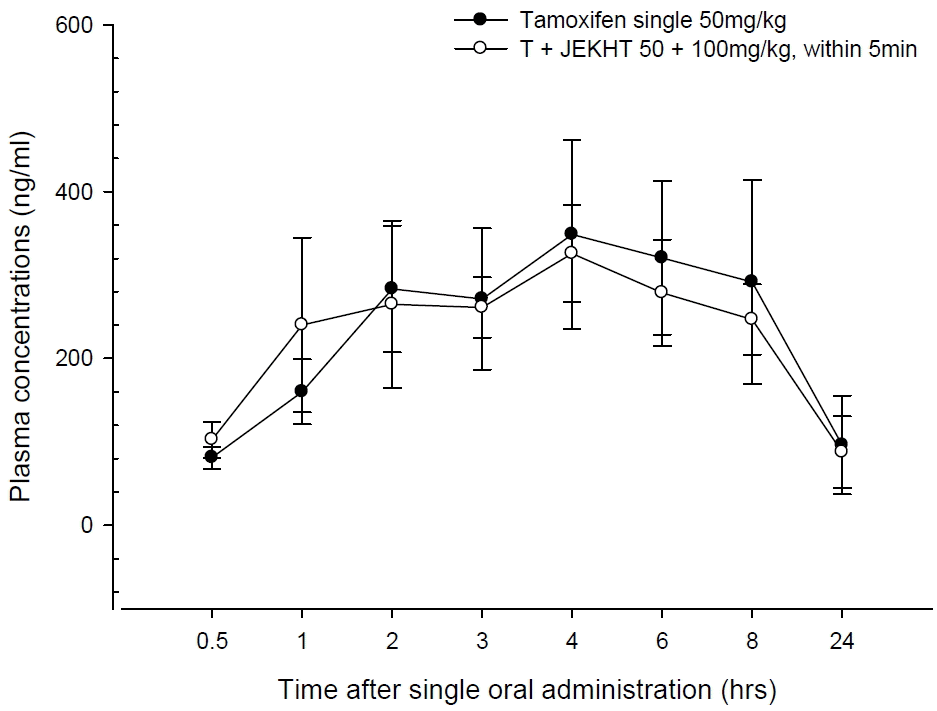
Plasma concentrations of tamoxifen with and without JEKHT co-administration in male rats. Tamoxifen was detected from 30 min to 24 hrs after end of administration in the both tamoxifen single and co-administered rats with JEKHT, respectively. No meaningful and no-significant changes on the plasma tamoxifen concentrations were detected in JEKHT co-administered rats as compared with as compared with tamoxifen single treated rats after single co-administration of tamoxifen 50 mg/kg with JEKHT 100 mg/kg, except for non-significant slight increases of plasma tamoxifen levels detected at 30 min and 1 hr after administration in JEKHT co-administered rats as compared with tamoxifen single treated rats, in the present study. Values are expressed as mean ± SD of five rats (ng/ml). T, tamoxifen, JEKHT: Jaeumkanghwatang aqueous extracts (Hanzung Pharm. Co., Daejeon, Korea).
2. Changes on the Tmax of tamoxifen
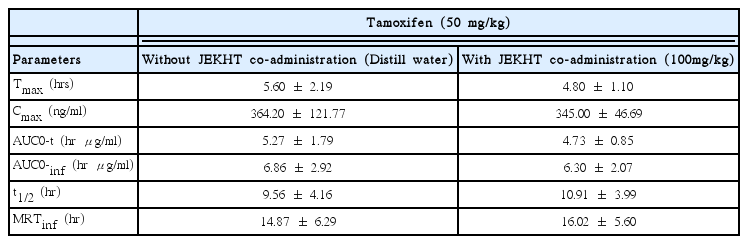
The Tmax of tamoxifen were slight and non-significantly decreased as −14.29% in co-administrated rats with tamoxifen 50 mg/kg and JEKHT 100 mg/kg (4.80±1.10 hr) as compared with tamoxifen single treated rats (5.60±2.19 hr), in the present study (Table 2).
Pharmacokinetic Parameters of Tamoxifen with and without JEKHT Co-Administration in Male Rats
3. Changes on the Cmax of tamoxifen
The Cmax of tamoxifen were slight and non-significantly decreased as -5.27% in co-administrated rats with tamoxifen 50 mg/kg and JEKHT 100 mg/kg (345.00±46.69 ng/ml) as compared with tamoxifen single treated rats (364.20±121.77 ng/ml), in the present study (Table 2).
4. Changes on the AUC of tamoxifen
The AUC0-t of tamoxifen were slight and non-significant decreased as −10.37% in co-administrated rats with tamoxifen 50 mg/kg and JEKHT 100 mg/kg (4.73±0.85 hr μg/ml) as compared with tamoxifen single treated rats (5.27±1.79 hr μg/ml). In addition, AUC0-inf of tamoxifen were also slight and non-significantly decreased as −8.16% in co-administrated rats with tamoxifen and JEKHT (6.30±2.07 hr μg/ml) as compared with tamoxifen single treated rats (6.86±2.92 hr μg/ml), in the present study (Table 2).
5. Changes on the t1/2 of tamoxifen
The t1/2 of tamoxifen were non-significantly increased as 14.16% in co-administrated rats with tamoxifen 50 mg/kg and JEKHT 100 mg/kg (10.91±3.99 hr) as compared with tamoxifen single treated rats (9.56±4.16 hr), in the present study (Table 2).
6. Changes on the MRTinf of tamoxifen
The MRTinf of tamoxifen were slight and non-significantly increased as 7.75% in co-administrated rats with tamoxifen 50 mg/kg and JEKHT 100 mg/kg (16.02±5.60 hr) as compared with tamoxifen single treated rats (14.87±6.29 hr), in the present study (Table 2).
Discussion
In the present study, the effects of JEKHT co-administration on the pharmacokinetics of tamoxifen were observed as a process of the comprehensive and integrative medicine, combination therapy of tamoxifen with JEKHT to achieve synergic pharmacodynamics and reduce toxicity on breast cancers. After 50 mg/kg of tamoxifen treatment, JEKHT 100 mg/kg was administered within 5 min. The plasma were collected at 30min before administration, 30 min, 1, 2, 3, 4, 6, 8 and 24 hrs after end of JEKHT treatment, and plasma concentrations of tamoxifen were analyzed using LC-MS/MS methods. PK parameters of tamoxifen (Tmax, Cmax, AUC, t1/2 and MRTinf) were analysis as compared with tamoxifen single administered. JEKHT did not influenced on the plasma concentrations and pharmacokinetics of tamoxifen after single oral co-administration, within 5 min except for some negligible effects on plasma concentration. The Tmax, Cmax, AUC, t1/2 and MRTinf of tamoxifen in co-administered rats were quite similar to those of tamoxifen single treated rats, respectively.
Tamoxifen was absorbed slowly following oral administration and Tmax of tamoxifen occur about 3–6 hrs after a single dose52–54) but it rapidly and extensively metabolized in the liver, through a substrate of CYP3A, 2C9, 2D626 including an active major metabolite, N-desmethyltamoxifen has biologic activity similar to that of the parent drug55,56). Steady-state concentrations of tamoxifen are attained after 3–4 weeks and those of N-desmethyltamoxifen, an active metabolite, are attained after 3–8 weeks57). Tamoxifen excreted principally in feces as polar conjugates58) with about 5–7 days of t1/2 in tamoxifen and 9–14 days in N-desmethyltamoxifen53). Clearance of tamoxifen is higher in female children 2–10 years of age than in women59,60). In the present study, Tmax of tamoxifen in tamoxifen single oral treated rats was detected as 5.60±2.19 hr, and Cmax, AUC0-t, AUC0-inf, t1/2 and MRTinf were detected as 364.20± 121.77 ng/ml, 5.27±1.79 hr μg/ml, 6.86±2.92 hr μ g/ml, 9.56±4.16 hr and 14.87±6.29 hr, respectively. In tamoxifen with JEKHT co-administered rats, Tmax, Cmax, AUC0-t, AUC0-inf, t1/2 and MRTinf of tamoxifen were detected as 4.80±1.10 hr, 345.00±46.69 ng/ml, 4.73±0.85 hr μg/ml, 6.30±2.07 hr μg/ml, 10.91± 3.99 hr and 16.02±5.60 hr; changed as −14.29, −5.27, −10.37, −8.16, 14.16 and 7.75% as compared with tamoxifen 50 mg/kg single oral treated rats. These results are considered as direct evidences that JEKHT did not influenced on the absorption and excretion of tamoxifen, when they were single orally co-administered within 5 min, at least.
Tamoxifen rapidly and extensively metabolized in the liver, through a substrate of CYP3A, 2C9, 2D626 to active major metabolite, N-desmethyltamoxifen55,56) and, therefore, tamoxifen can be interacted with various drugs like aminoglutethimide34), anticoagulants 35,36), bromocriptine37), letrozole38), medroxyprogesterone39), phenobarbital40) and rifampin41). In addition the possibilities that tamoxifen competitively interacted with cyclosporine, erythromycin, diltiazem, erythromycin and nifedipine were also suggested in vitro experiments42–44). The severities of various side effects arise from tamoxifen treatment, especially bone loss19), endometrial cancer20), thromboembolism 21), fatty liver22), reduced cognition23), semantic memory scores24) and libido25,26), premature growth plate fusion27), immune suppression28,29) and hypersensitivity30,31) are considered as directly co-related with absorption and excretion of tamoxifen or pharmacodynamics. In the present study, co-administration of JEKHT did not influenced on the plasma concentrations and pharmacokinetics of tamoxifen. It means that JEKHT didn’t interact with tamoxifen.
Based on the results of the present study, JEKHT did not influenced on the oral bioavailability of tamoxifen, when they were single co-administered within 5 min. However, more detail pharmacokinetic studies should be tested to conclude the possibilities that can be used as comprehensive and integrative therapy with JEKHT and tamoxifen for breast cancers, when they were co-administered, like the effects on the pretreatment of JEKHT and after repeat co-administrations.
Acknowledgments
This study was supported by grant of Korea of Health & Welfare, Republic of Korea (Project No: 20-11-0-090-091-3000-3033-320).
Notes
Conflicts of interest
The authors declare that there is no conflict of interest regarding the publication of this paper.